Daliresp: Package Insert / Prescribing Info
Package insert / product label
Generic name: roflumilast
Dosage form: tablet
Drug class: Selective phosphodiesterase-4 inhibitors
Medically reviewed by Drugs.com. Last updated on Sep 9, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
DALIRESP® (roflumilast) tablets
Initial U.S. Approval: 2011
Indications and Usage for Daliresp
Daliresp Dosage and Administration
The maintenance dose for patients with COPD is one 500 mcg tablet per day, with or without food. Starting treatment with a dose of DALIRESP 250 mcg once daily for 4 weeks and increasing to DALIRESP 500 mcg once daily thereafter may reduce the rate of treatment discontinuation in some patients. (2)
Dosage Forms and Strengths
Tablets: 250 mcg, 500 mcg (3)
Contraindications
Moderate to severe liver impairment (Child-Pugh B or C) (4)
Warnings and Precautions
- •
- Acute Bronchospasm: Do not use for the relief of acute bronchospasm. (5.1)
- •
- Psychiatric Events including Suicidality: Advise patients, their caregivers, and families to be alert for the emergence or worsening of insomnia, anxiety, depression, suicidal thoughts or other mood changes, and if such changes occur to contact their healthcare provider. Carefully weigh the risks and benefits of treatment with DALIRESP in patients with a history of depression and/or suicidal thoughts or behavior. (5.2)
- •
- Weight Decrease: Monitor weight regularly. If unexplained or clinically significant weight loss occurs, evaluate weight loss and consider discontinuation of DALIRESP. (5.3)
- •
- Drug Interactions: Use with strong cytochrome P450 enzyme inducers (e.g., rifampicin, phenobarbital, carbamazepine, phenytoin) is not recommended. (5.4)
Adverse Reactions/Side Effects
Most common adverse reactions (≥2%) are diarrhea, weight decrease, nausea, headache, back pain, influenza, insomnia, dizziness and decreased appetite. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Use with inhibitors of CYP3A4 or dual inhibitors of CYP3A4 and CYP1A2 (e.g., erythromycin, ketoconazole, fluvoxamine, enoxacin, cimetidine) will increase roflumilast systemic exposure and may result in increased adverse reactions. The risk of such concurrent use should be weighed carefully against benefit. (7.2)
Use In Specific Populations
Nursing Mothers: DALIRESP should not be used by women who are nursing as excretion of roflumilast and/or its metabolites into human milk is probable and there are no human studies that have investigated effects of DALIRESP on breast-fed infants. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2019
Full Prescribing Information
1. Indications and Usage for Daliresp
DALIRESP® is indicated as a treatment to reduce the risk of COPD exacerbations in patients with severe COPD associated with chronic bronchitis and a history of exacerbations.
2. Daliresp Dosage and Administration
The maintenance dose of DALIRESP is one 500 micrograms (mcg) tablet per day, with or without food.
Starting treatment with a dose of DALIRESP 250 mcg once daily for 4 weeks and increasing to DALIRESP 500 mcg once daily thereafter may reduce the rate of treatment discontinuation in some patients [see Clinical Studies (14.1)]. However, 250 mcg per day is not the effective (therapeutic) dose.
3. Dosage Forms and Strengths
- •
- DALIRESP 250 mcg tablets are white to off-white, round, embossed with “D” on one side and “250” on the other side
- •
- DALIRESP 500 mcg tablets are white to off-white, round, embossed with “D” on one side and “500” on the other side
4. Contraindications
The use of DALIRESP is contraindicated in the following condition:
Moderate to severe liver impairment (Child-Pugh B or C) [see Clinical Pharmacology (12.3) and Use in Specific Populations (8.6)].
5. Warnings and Precautions
5.1 Treatment of Acute Bronchospasm
DALIRESP is not a bronchodilator and should not be used for the relief of acute bronchospasm.
5.2 Psychiatric Events including Suicidality
Treatment with DALIRESP is associated with an increase in psychiatric adverse reactions. In 8 controlled clinical trials 5.9% (263) of patients treated with DALIRESP 500 mcg daily reported psychiatric adverse reactions compared to 3.3% (137) treated with placebo. The most commonly reported psychiatric adverse reactions were insomnia, anxiety, and depression which were reported at higher rates in those treated with DALIRESP 500 mcg daily (2.4%, 1.4%, and 1.2% for DALIRESP versus 1.0%, 0.9%, and 0.9% for placebo, respectively) [see Adverse Reactions (6.1)]. Instances of suicidal ideation and behavior, including completed suicide, have been observed in clinical trials. Three patients experienced suicide-related adverse reactions (one completed suicide and two suicide attempts) while receiving DALIRESP compared to one patient (suicidal ideation) who received placebo. One patient completed suicide while receiving DALIRESP in Trial 9 [see Clinical Studies (14.1)], which assessed the effect of adding roflumilast to a fixed-dose combination (FDC) of ICS/LABA on rates of exacerbations in COPD patients over 1 year of treatment. Cases of suicidal ideation and behavior, including completed suicide, have been observed in the post-marketing setting in patients with or without a history of depression.
Before using DALIRESP in patients with a history of depression and/or suicidal thoughts or behavior, prescribers should carefully weigh the risks and benefits of treatment with DALIRESP in such patients. Patients, their caregivers, and families should be advised of the need to be alert for the emergence or worsening of insomnia, anxiety, depression, suicidal thoughts or other mood changes, and if such changes occur to contact their healthcare provider. Prescribers should carefully evaluate the risks and benefits of continuing treatment with DALIRESP if such events occur.
5.3 Weight Decrease
Weight loss was a common adverse reaction in DALIRESP clinical trials and was reported in 7.5% (331) of patients treated with DALIRESP 500 mcg once daily compared to 2.1% (89) treated with placebo [see Adverse Reactions (6.1)]. In addition to being reported as adverse reactions, weight was prospectively assessed in two placebo-controlled clinical trials of one year duration. In these studies, 20% of patients receiving roflumilast experienced moderate weight loss (defined as between 5-10% of body weight) compared to 7% of patients who received placebo. In addition, 7% of patients who received roflumilast compared to 2% of patients receiving placebo experienced severe (>10% body weight) weight loss. During follow-up after treatment discontinuation, the majority of patients with weight loss regained some of the weight they had lost while receiving DALIRESP. Patients treated with DALIRESP should have their weight monitored regularly. If unexplained or clinically significant weight loss occurs, weight loss should be evaluated, and discontinuation of DALIRESP should be considered.
5.4 Drug Interactions
A major step in roflumilast metabolism is the N-oxidation of roflumilast to roflumilast N-oxide by CYP3A4 and CYP1A2. The administration of the cytochrome P450 enzyme inducer rifampicin resulted in a reduction in exposure, which may result in a decrease in the therapeutic effectiveness of DALIRESP. Therefore, the use of strong cytochrome P450 enzyme inducers (e.g., rifampicin, phenobarbital, carbamazepine, phenytoin) with DALIRESP is not recommended [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
6. Adverse Reactions/Side Effects
The following adverse reactions are described in greater detail in other sections:
- •
- Psychiatric Events Including Suicidality [see Warnings and Precautions (5.2)]
- •
- Weight Decrease [see Warnings and Precautions (5.3)]
6.1 Adverse Reactions in Clinical Studies
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described below reflect exposure of 4438 patients to DALIRESP 500 mcg once daily in four 1-year placebo-controlled trials, two 6-month placebo-controlled trials, and two 6-month drug add-on trials [see Clinical Studies (14.1)]. In these trials, 3136 and 1232 COPD patients were exposed to DALIRESP 500 mcg once daily for 6 months and 1 year, respectively.
The population had a median age of 64 years (range 40-91), 73% were male, 92.9% were Caucasian, and had COPD with a mean pre-bronchodilator forced expiratory volume in one second (FEV1) of 8.9 to 89.1% predicted. In these trials, 68.5% of the patients treated with DALIRESP reported an adverse reaction compared with 65.3% treated with placebo.
The proportion of patients who discontinued treatment due to adverse reaction was 14.8% for DALIRESP-treated patients and 9.9% for placebo-treated patients. The most common adverse reactions that led to discontinuation of DALIRESP were diarrhea (2.4%) and nausea (1.6%).
Serious adverse reactions, whether considered drug-related or not by the investigators, which occurred more frequently in DALIRESP-treated patients include diarrhea, atrial fibrillation, lung cancer, prostate cancer, acute pancreatitis, and acute renal failure.
Table 1 summarizes the adverse reactions reported by ≥2% of patients in the DALIRESP group in 8 controlled COPD clinical trials.
|
Treatment |
||
|
Adverse Reactions |
DALIRESP |
Placebo |
|
(Preferred Term) |
(N=4438) |
(N=4192) |
|
n (%) |
n (%) |
|
|
Diarrhea |
420 (9.5) |
113 (2.7) |
|
Weight decreased |
331 (7.5) |
89 (2.1) |
|
Nausea |
209 (4.7) |
60 (1.4) |
|
Headache |
195 (4.4) |
87 (2.1) |
|
Back pain |
142 (3.2) |
92 (2.2) |
|
Influenza |
124 (2.8) |
112 (2.7) |
|
Insomnia |
105 (2.4) |
41 (1.0) |
|
Dizziness |
92 (2.1) |
45 (1.1) |
|
Decreased appetite |
91 (2.1) |
15 (0.4) |
Adverse reactions that occurred in the DALIRESP group at a frequency of 1 to 2% where rates exceeded that in the placebo group include:
Gastrointestinal disorders - abdominal pain, dyspepsia, gastritis, vomiting
Infections and infestations - rhinitis, sinusitis, urinary tract infection
Musculoskeletal and connective tissue disorders - muscle spasms
Nervous system disorders - tremor
Psychiatric disorders - anxiety, depression
The safety profile of roflumilast reported during Trial 9 was consistent with the key pivotal studies.
6.2 Postmarketing Experience
The following adverse reactions have been identified from spontaneous reports of DALIRESP received worldwide and have not been listed elsewhere. These adverse reactions have been chosen for inclusion due to a combination of seriousness, frequency of reporting or potential causal connection to DALIRESP. Because these adverse reactions were reported voluntarily from a population of uncertain size, it is not possible to estimate their frequency or establish a causal relationship to DALIRESP exposure: hypersensitivity reactions (including angioedema, urticaria, and rash), gynecomastia.
7. Drug Interactions
A major step in roflumilast metabolism is the N-oxidation of roflumilast to roflumilast N-oxide by CYP3A4 and CYP1A2 [see Clinical Pharmacology (12.3)].
7.1 Drugs that Induce Cytochrome P450 (CYP) Enzymes
Strong cytochrome P450 enzyme inducers decrease systemic exposure to roflumilast and may reduce the therapeutic effectiveness of DALIRESP. Therefore the use of strong cytochrome P450 inducers (e.g., rifampicin, phenobarbital, carbamazepine, and phenytoin) with DALIRESP is not recommended [see Warnings and Precautions (5.4) and Clinical Pharmacology (12.3)].
7.2 Drugs that Inhibit Cytochrome P450 (CYP) Enzymes
The co-administration of DALIRESP (500 mcg) with CYP3A4 inhibitors or dual inhibitors that inhibit both CYP3A4 and CYP1A2 simultaneously (e.g., erythromycin, ketoconazole, fluvoxamine, enoxacin, cimetidine) may increase roflumilast systemic exposure and may result in increased adverse reactions. The risk of such concurrent use should be weighed carefully against benefit [see Clinical Pharmacology (12.3)].
7.3 Oral Contraceptives Containing Gestodene and Ethinyl Estradiol
The co-administration of DALIRESP (500 mcg) with oral contraceptives containing gestodene and ethinyl estradiol may increase roflumilast systemic exposure and may result in increased side effects. The risk of such concurrent use should be weighed carefully against benefit [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no randomized clinical studies of DALIRESP in pregnant women. In animal reproductive toxicity studies, DALIRESP administered to pregnant rats and rabbits during the period of organogenesis produced no fetal structural abnormalities. The highest DALIRESP dose in these studies was approximately 30 and 26 times, respectively, the maximum recommended human dose (MRHD). DALIRESP induced post-implantation loss in rats at doses greater than or equal to approximately 10 times the MRHD. DALIRESP induced stillbirth and decreased pup viability in mice at doses corresponding to approximately 16 and 49 times, respectively, the MRHD. DALIRESP has been shown to adversely affect pup post-natal development when dams were treated with the drug during pregnancy and lactation periods in mice at doses corresponding to 49 times the MRHD (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Labor and delivery
DALIRESP should not be used during labor and delivery. There are no human studies that have investigated effects of DALIRESP on preterm labor or labor at term; however, animal studies showed that DALIRESP disrupted the labor and delivery process in mice.
Data
Animal data
In an embryo-fetal development study, pregnant rats were dosed orally during the period of organogenesis with up to 1.8 mg/kg/day DALIRESP (approximately 30 times the MRHD on an AUC basis). No evidence of structural abnormalities or effects on survival rates were observed. DALIRESP did not affect embryo-fetal development at approximately 3 times the MRHD (on a mg/m2 basis at a maternal oral dose of 0.2 mg/kg/day).
In a fertility and embryo-fetal development study, male rats were dosed orally with up to 1.8 mg/kg/day DALIRESP for 10 weeks and females for two weeks prior to pairing and throughout the organogenesis period. DALIRESP induced pre- and post-implantation loss at doses greater than or equal to approximately 10 times the MRHD (on a mg/m2 basis at maternal oral doses greater than or equal to 0.6 mg/kg/day). DALIRESP did not cause fetal structural abnormalities at exposures up to approximately 29 times the MRHD (on an AUC basis at maternal oral doses up to 1.8 mg/kg/day).
In an embryo-fetal development study in rabbits, pregnant does were dosed orally with 0.8 mg/kg/day DALIRESP during the period of organogenesis. DALIRESP did not cause fetal structural abnormalities at exposures approximately 26 times the MRHD (on a mg/m2 basis at maternal oral doses of 0.8 mg/kg/day).
In pre- and post-natal developmental studies in mice, dams were dosed orally with up to 12 mg/kg/day
DALIRESP during the period of organogenesis and lactation. DALIRESP induced stillbirth and decreased pup
viability at doses corresponding to approximately 16 and 49 times, respectively, the MRHD (on a mg/m2
basis at maternal doses >2 mg/kg/day and 6 mg/kg/day, respectively). DALIRESP induced delivery retardation in pregnant mice at doses greater or equal to approximately 16 times the MRHD (on a mg/m2 basis at maternal doses >2 mg/kg/day). DALIRESP decreased pup rearing frequencies at approximately 49 times the MRHD (on a mg/m2 basis at a maternal dose of 6 mg/kg/day) during pregnancy and lactation. DALIRESP also decreased survival and forelimb grip reflex and delayed pinna detachment in mouse pups at approximately 97 times the MRHD (on a mg/m2 basis at a maternal dose of 12 mg/kg/day).
8.2 Lactation
Risk Summary
There is no information regarding the presence of DALIRESP in human milk, the effects on the breastfed infant, or the effects on milk production.
Roflumilast and/or its metabolites are excreted into the milk of lactating rats. Excretion of roflumilast and/or its metabolites into human milk is probable. DALIRESP should not be used by women who are nursing.
Data
Animal data
Roflumilast and/or its metabolite concentrations measured 8 hours after an oral dose of 1 mg/kg given to lactating rats were 0.32 and 0.02 mcg/g in the milk and pup liver, respectively.
8.4 Pediatric Use
COPD does not normally occur in children. The safety and effectiveness of DALIRESP in pediatric patients have not been established.
8.5 Geriatric Use
Of the 4438 COPD subjects exposed to DALIRESP for up to 12 months in 8 controlled clinical trials, 2022 were >65 years of age and 471 were >75 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Based on available data for roflumilast, no adjustment of dosage in geriatric patients is warranted [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
Roflumilast 250 mcg once daily for 14 days was studied in subjects with mild-to-moderate hepatic impairment classified as Child-Pugh A and B (8 subjects in each group). The AUCs of roflumilast and roflumilast N-oxide were increased by 51% and 24%, respectively, in Child-Pugh A subjects and by 92% and 41%, respectively, in Child-Pugh B subjects, as compared to age-, weight-, and gender-matched healthy subjects. The Cmax of roflumilast and roflumilast N-oxide were increased by 3% and 26%, respectively in Child-Pugh A subjects and by 26% and 40%, respectively in Child-Pugh B subjects, as compared to healthy subjects. DALIRESP 500 mcg has not been studied in hepatically impaired patients. Clinicians should consider the risk-benefit of administering DALIRESP to patients who have mild liver impairment (Child-Pugh A). DALIRESP is not recommended for use in patients with moderate or severe liver impairment (Child-Pugh B or C) [see Contraindications (4) and Clinical Pharmacology (12.3)].
8.7 Renal Impairment
In twelve subjects with severe renal impairment administered a single dose of 500 mcg roflumilast, the AUCs of roflumilast and roflumilast N-oxide were decreased by 21% and 7%, respectively and Cmax were reduced by 16% and 12%, respectively. No dosage adjustment is necessary for patients with renal impairment [see Clinical Pharmacology (12.3)].
10. Overdosage
10.1 Human Experience
No case of overdose has been reported in clinical studies with DALIRESP. During the Phase I studies of DALIRESP, the following symptoms were observed at an increased rate after a single oral dose of 2500 mcg and a single dose of 5000 mcg: headache, gastrointestinal disorders, dizziness, palpitations, lightheadedness, clamminess, and arterial hypotension.
10.2 Management of Overdose
In case of overdose, patients should seek immediate medical help. Appropriate supportive medical care should be provided. Since roflumilast is highly protein bound, hemodialysis is not likely to be an efficient method of drug removal. It is not known whether roflumilast is dialyzable by peritoneal dialysis.
11. Daliresp Description
The active ingredient in DALIRESP tablets is roflumilast. Roflumilast and its active metabolite (roflumilast N-oxide) are selective phosphodiesterase 4 (PDE4) inhibitors. The chemical name of roflumilast is N-(3,5-dichloropyridin-4-yl)-3-cyclopropylmethoxy-4-difluoromethoxy-benzamide. Its empirical formula is C17H14Cl2F2N2O3 and the molecular weight is 403.22.
The chemical structure is:
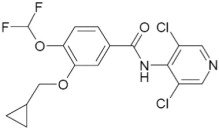
The drug substance is a white to off-white non-hygroscopic powder with a melting point of 160°C. It is practically insoluble in water and hexane, sparingly soluble in ethanol, and freely soluble in acetone.
DALIRESP is supplied as white to off-white, round tablets, embossed with “D” on one side and “250” or “500” on the other side. Each tablet contains 250 mcg or 500 mcg of roflumilast.
Each tablet of DALIRESP for oral administration contains the following inactive ingredients: lactose monohydrate, corn starch, povidone, and magnesium stearate.
12. Daliresp - Clinical Pharmacology
12.1 Mechanism of Action
Roflumilast and its active metabolite (roflumilast N-oxide) are selective inhibitors of phosphodiesterase 4 (PDE4). Roflumilast and roflumilast N-oxide inhibition of PDE4 (a major cyclic-3′,5′-adenosine monophosphate (cyclic AMP)-metabolizing enzyme in lung tissue) activity leads to accumulation of intracellular cyclic AMP. While the specific mechanism(s) by which DALIRESP exerts its therapeutic action in COPD patients is not well defined, it is thought to be related to the effects of increased intracellular cyclic AMP in lung cells.
12.2 Pharmacodynamics
In COPD patients, 4‑week treatment with DALIRESP 500 mcg oral once daily reduced sputum neutrophils and eosinophils by 31%, and 42%, respectively. In a pharmacodynamic study in healthy volunteers, DALIRESP 500 mcg once daily reduced the number of total cells, neutrophils and eosinophils found in bronchoalveolar lavage fluid following segmental pulmonary lipopolysaccharide (LPS) challenge by 35%, 38% and 73%, respectively. The clinical significance of these findings is unknown.
12.3 Pharmacokinetics
Absorption
The absolute bioavailability of roflumilast following a 500 mcg oral dose is approximately 80%. Maximum plasma concentrations (Cmax) of roflumilast typically occur approximately one hour after dosing (ranging from 0.5 to 2 hours) in the fasted state while plateau-like maximum concentrations of the N-oxide metabolite are reached in approximately eight hours (ranging from 4 to 13 hours). Food has no effect on total drug absorption, but delays time to maximum concentration (Tmax) of roflumilast by one hour and reduces Cmax by approximately 40%, however, Cmax and Tmax of roflumilast N-oxide are unaffected. An in vitro study showed that roflumilast and roflumilast N-oxide did not inhibit P-gp transporter.
Distribution
Plasma protein binding of roflumilast and its N-oxide metabolite is approximately 99% and 97%, respectively. Volume of distribution for single‑dose 500 mcg roflumilast is about 2.9 L/kg. Studies in rats with radiolabeled roflumilast indicate low penetration across the blood-brain barrier.
Metabolism
Roflumilast is extensively metabolized via Phase I (cytochrome P450) and Phase II (conjugation) reactions. The N-oxide metabolite is the only major metabolite observed in the plasma of humans. Together, roflumilast and roflumilast N-oxide account for the majority (87.5%) of total dose administered in plasma. In urine, roflumilast was not detectable while roflumilast N-oxide was only a trace metabolite (less than 1%). Other conjugated metabolites such as roflumilast N-oxide glucuronide and 4-amino-3,5-dichloropyridine N-oxide were detected in urine.
While roflumilast is three times more potent than roflumilast N-oxide at inhibition of the PDE4 enzyme in vitro, the plasma AUC of roflumilast N-oxide on average is about 10-fold greater than the plasma AUC of roflumilast.
In vitro studies and clinical drug-drug interaction studies suggest that the biotransformation of roflumilast to its N-oxide metabolite is mediated by CYP1A2 and 3A4. Based on further in vitro results in human liver microsomes, therapeutic plasma concentrations of roflumilast and roflumilast N-oxide do not inhibit CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5, or 4A9/11. Therefore, there is a low probability of relevant interactions with substances metabolized by these P450 enzymes. In addition, in vitro studies demonstrated no induction of the CYP1A2, 2A6, 2C9, 2C19, or 3A4/5 and only a weak induction of CYP2B6 by roflumilast.
Elimination
The plasma clearance after short-term intravenous infusion of roflumilast is on average about 9.6 L/h. Following an oral dose, the median plasma effective half-life of roflumilast and its N-oxide metabolite are approximately 17 and 30 hours, respectively. Steady state plasma concentrations of roflumilast and its N-oxide metabolite are reached after approximately 4 days for roflumilast and 6 days for roflumilast N-oxide following once‑daily dosing. Following intravenous or oral administration of radiolabeled roflumilast, about 70% of the radioactivity was recovered in the urine.
Special Populations
Hepatic Impairment
Roflumilast 250 mcg once daily for 14 days was studied in subjects with mild-to-moderate hepatic impairment classified as Child-Pugh A and B (8 subjects in each group). The AUC of roflumilast and roflumilast N-oxide were increased by 51% and 24%, respectively in Child-Pugh A subjects and by 92% and 41%, respectively, in Child-Pugh B subjects, as compared to age-, weight-, and gender-matched healthy subjects. The Cmax of roflumilast and roflumilast N-oxide were increased by 3% and 26%, respectively, in Child-Pugh A subjects and by 26% and 40%, respectively in Child-Pugh B subjects, as compared to healthy subjects. DALIRESP 500 mcg has not been studied in hepatically impaired patients. Clinicians should consider the risk-benefit of administering DALIRESP to patients who have mild liver impairment (Child-Pugh A). DALIRESP is not recommended for use in patients with moderate or severe liver impairment (Child-Pugh B or C) [see Contraindications (4) and Use in Specific Populations (8.6)].
Renal Impairment
In twelve subjects with severe renal impairment administered a single dose of 500 mcg roflumilast, roflumilast and roflumilast N-oxide AUCs were decreased by 21% and 7%, respectively and Cmax were reduced by 16% and 12%, respectively. No dosage adjustment is necessary for patients with renal impairment [see Use in Specific Populations (8.7)].
Age
Roflumilast 500 mcg once daily for 15 days was studied in young, middle aged, and elderly healthy subjects. The exposure in elderly (>65 years of age) were 27% higher in AUC and 16% higher in Cmax for roflumilast and 19% higher in AUC and 13% higher in Cmax for roflumilast-N-oxide than that in young volunteers (18-45 years old). No dosage adjustment is necessary for elderly patients [see Use in Specific Populations (8.5)].
Gender
In a Phase I study evaluating the effect of age and gender on the pharmacokinetics of roflumilast and roflumilast N-oxide, a 39% and 33% increase in roflumilast and roflumilast N-oxide AUC were noted in healthy female subjects as compared to healthy male subjects. No dosage adjustment is necessary based on gender.
Smoking
The pharmacokinetics of roflumilast and roflumilast N-oxide were comparable in smokers as compared to non-smokers. There was no difference in Cmax between smokers and non-smokers when roflumilast 500 mcg was administered as a single dose to 12 smokers and 12 non-smokers. The AUC of roflumilast in smokers was 13% less than that in non-smokers while the AUC of roflumilast N-oxide in smokers was 17% more than that in non-smokers.
Race
As compared to Caucasians, African Americans, Hispanics, and Japanese showed 16%, 41%, and 15% higher AUC, respectively, for roflumilast and 43%, 27%, and 16% higher AUC, respectively, for roflumilast N-oxide. As compared to Caucasians, African Americans, Hispanics, and Japanese showed 8%, 21%, and 5% higher Cmax, respectively, for roflumilast and 43%, 27%, and 17% higher Cmax, respectively, for roflumilast N-oxide. No dosage adjustment is necessary for race.
Drug Interactions
Drug interaction studies were performed with roflumilast and other drugs likely to be coadministered or drugs commonly used as probes for pharmacokinetic interaction [see Drug Interactions (7)]. No significant drug interactions were observed when 500 mcg oral roflumilast was administered with inhaled salbutamol, formoterol, budesonide and oral montelukast, digoxin, theophylline, warfarin, sildenafil, midazolam, or antacids.
The effect of concomitant drugs on the exposure of roflumilast and roflumilast N-oxide is shown in the Figure 1 below.
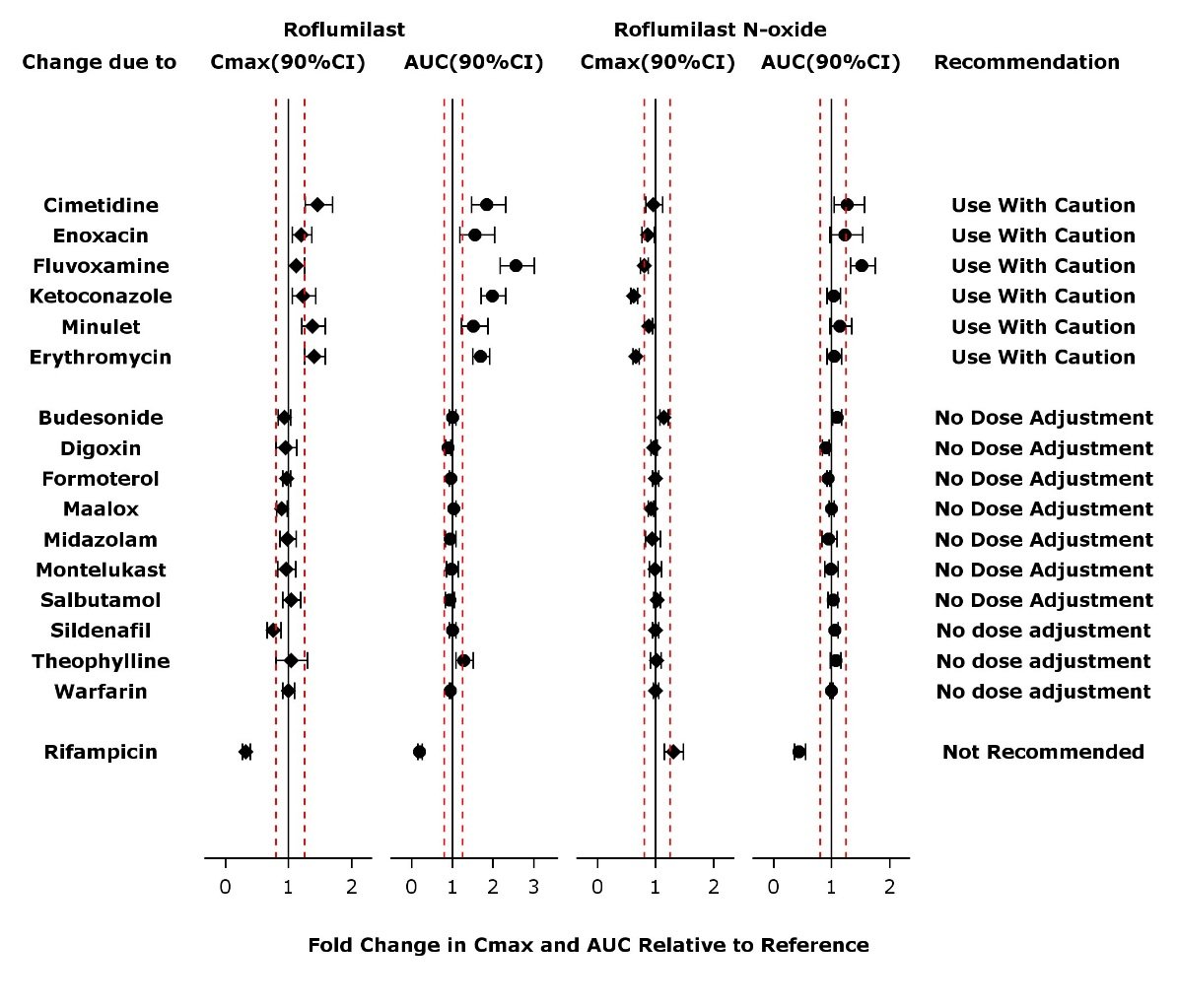
Figure 1. Effect of concomitant drugs on the exposure of roflumilast and roflumilast N-oxide. Note that the dashed lines indicate the lower and higher bounds (0.8-1.25) of the 90% confidence interval of the geometric mean ratio of Cmax or AUC for roflumilast or roflumilast N-oxide for Treatment (DALIRESP+Coadministered Drug) vs. Reference (DALIRESP). The dosing regimens of coadministered drugs was: Midazolam: 2 mg po SD; Erythromycin: 500 mg po TID; Ketoconazole: 200 mg po BID; Rifampicin: 600 mg po QD; Fluvoxamine: 50 mg po QD; Digoxin: 250 mcg po SD; Maalox: 30 mL po SD; Salbutamol: 0.2 mg po TID; Cimetidine: 400 mg po BID; Formoterol: 40 mcg po BID; Budesonide: 400 mcg po BID; Theophylline: 375 mg po BID; Warfarin: 250 mg po SD; Enoxacin: 400 mg po BID; Sildenafil: 100 mg SD; Minulet (combination oral contraceptive): 0.075 mg gestodene/0.03 mg ethinylestradiol po QD; Montelukast: 10 mg po QD
Drug interactions considered to be significant are described in more detail below [see Warnings and Precautions (5.4) and Drug Interactions (7)].
Inhibitors of CYP3A4 and CYP1A2:
Erythromycin: In an open-label crossover study in 16 healthy volunteers, the coadministration of CYP3A4 inhibitor erythromycin (500 mg three times daily for 13 days) with a single oral dose of 500 mcg DALIRESP resulted in 40% and 70% increase in Cmax and AUC for roflumilast, respectively, and a 34% decrease and a 4% increase in Cmax and AUC for roflumilast N-oxide, respectively.
Ketoconazole: In an open-label crossover study in 16 healthy volunteers, the coadministration of a strong CYP3A4 inhibitor ketoconazole (200 mg twice daily for 13 days) with a single oral dose of 500 mcg DALIRESP resulted in 23% and 99% increase in Cmax and AUC for roflumilast, respectively, and a 38% reduction and 3% increase in Cmax and AUC for roflumilast N-oxide, respectively.
Fluvoxamine: In an open-label crossover study in 16 healthy volunteers, the coadministration of dual CYP 3A4/1A2 inhibitor fluvoxamine (50 mg daily for 14 days) with a single oral dose of 500 mcg DALIRESP showed a 12% and 156% increase in roflumilast Cmax and AUC along with a 210% decrease and 52% increase in roflumilast N-oxide Cmax and AUC, respectively.
Enoxacin: In an open-label crossover study in 16 healthy volunteers, the coadministration of dual CYP 3A4/1A2 inhibitor enoxacin (400 mg twice daily for 12 days) with a single oral dose of 500 mcg DALIRESP resulted in an increased Cmax and AUC of roflumilast by 20% and 56%, respectively. Roflumilast N-oxide Cmax was decreased by 14% while roflumilast N-oxide AUC was increased by 23%.
Cimetidine: In an open-label crossover study in 16 healthy volunteers, the coadministration of a dual CYP 3A4/1A2 inhibitor cimetidine (400 mg twice daily for 7 days) with a single dose of 500 mcg oral DALIRESP resulted in a 46% and 85% increase in roflumilast Cmax and AUC; and a 4% decrease in Cmax and 27% increase in AUC for roflumilast N-oxide, respectively.
Oral Contraceptives containing Gestodene and Ethinyl Estradiol:
In an open-label crossover study in 20 healthy adult volunteers, coadministration of a single oral dose of 500 mcg DALIRESP with repeated doses of a fixed combination oral contraceptive containing 0.075 mg gestodene and 0.03 mg ethinyl estradiol to steady state caused a 38% increase and 12% decrease in Cmax of roflumilast and roflumilast N-oxide, respectively. Roflumilast and roflumilast N-oxide AUCs were increased by 51% and 14%, respectively.
Inducers of CYP enzymes:
Rifampicin: In an open-label, three-period, fixed-sequence study in 15 healthy volunteers, coadministration of the strong CYP3A4 inducer rifampicin (600 mg once daily for 11 days) with a single oral dose of 500 mcg DALIRESP resulted in reduction of roflumilast Cmax and AUC by 68% and 79%, respectively; and an increase of roflumilast N-oxide Cmax by 30% and reduced roflumilast N-oxide AUC by 56%.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies were conducted in hamsters and mice with roflumilast to evaluate its carcinogenic potential. In 2-year oral gavage carcinogenicity studies, roflumilast treatment resulted in dose-related, statistically significant increases in the incidence of undifferentiated carcinomas of nasal epithelium in hamsters at ≥8 mg/kg/day (approximately 11 times the MRHD based on summed AUCs of roflumilast and its metabolites). The tumorigenicity of roflumilast appears to be attributed to a reactive metabolite of 4-amino-3,5-dichloro-pyridine N-oxide (ADCP N-oxide). No evidence of tumorigenicity was observed in mice at roflumilast oral doses up to 12 and 18 mg/kg/day in females and males, respectively (approximately 10 and 15 times the MRHD, respectively, based on summed AUCs of roflumilast and its metabolites).
Roflumilast tested positive in an in vivo mouse micronucleus test, but negative in the following assays: Ames test for bacterial gene mutation, in vitro chromosome aberration assay in human lymphocytes, in vitro HPRT test with V79 cells, an in vitro micronucleus test with V79 cells, DNA adduct formation assay in rat nasal mucosa, liver and testes, and in vivo mouse bone marrow chromosome aberration assay. Roflumilast N-oxide was negative in the Ames test and in vitro micronucleus test with V79 cells.
In a human spermatogenesis study, roflumilast 500 mcg had no effects on semen parameters or reproductive hormones during the 3-month treatment period and the following 3-month off-treatment period. In a fertility study, roflumilast decreased fertility rates in male rats at 1.8 mg/kg/day (approximately 29 times the MRHD on a mg/m2 basis). The male rats also showed increases in the incidence of tubular atrophy, degeneration in the testis and spermiogenic granuloma in the epididymides. No effect on rat fertility rate or male reproductive organ morphology was observed at 0.6 mg/kg/day (approximately 10 times the MRHD on a mg/m2 basis). In a female fertility study, no effect on fertility was observed up to the highest roflumilast dose of 1.5 mg/kg/day in rats (approximately 24 times the MRHD on a mg/m2 basis).
14. Clinical Studies
14.1 Chronic Obstructive Pulmonary Disease (COPD)
The efficacy and safety of DALIRESP (roflumilast) in COPD was evaluated in 8 randomized, double-blind, controlled, parallel‑group clinical trials in 9394 adult patients (4425 receiving DALIRESP 500 mcg) 40 years of age and older with COPD. Of the 8 trials, two were placebo-controlled dose selection trials (Trials 1 and 2) of 6 months’ duration that evaluated the efficacy of DALIRESP 250 mcg and 500 mcg once daily, four were placebo-controlled 1-year trials (Trials 3, 4, 5, and 6) primarily designed to evaluate the efficacy of DALIRESP on COPD exacerbations, and two were 6-month efficacy trials (Trials 7 and 8) which assessed the effect of DALIRESP as add-on therapy to a long-acting beta agonist or long-acting anti-muscarinic. The 8 trials enrolled patients with nonreversible obstructive lung disease (FEV1/FVC ≤70% and ≤12% or 200 mL improvement in FEV1 in response to 4 puffs of albuterol/salbutamol) but the severity of airflow obstruction at baseline was different among the trials. Patients enrolled in the dose selection trials had the full range of COPD severity (FEV1 30-80% predicted); median age of 63 years, 73% male, and 99% Caucasian. Patients enrolled in the four exacerbation trials had severe COPD (FEV1 ≤50% predicted); median age of 64 years, 74% male, and 90% Caucasian.
Patients enrolled in the two 6-month efficacy trials had moderate to severe COPD (FEV1 40-70% predicted); median age of 65 years, 68% male, and 97% Caucasian. COPD exacerbations and lung function (FEV1) were co-primary efficacy outcome measures in the four 1-year trials. In the two 6-month supportive efficacy trials, lung function (FEV1) alone was the primary efficacy outcome measure.
The two 6-month dose-selection efficacy trials (Trials 1 and 2) explored doses of 250 mcg and 500 mcg once daily in a total of 1929 patients (751 and 724 on DALIRESP 250 and 500 mcg, respectively). The selection of the 500 mcg dose was primarily based on nominal improvements in lung function (FEV1) over the 250 mcg dose. The once‑daily dosing regimen was primarily based on the determination of a plasma half-life of 17 hours for roflumilast and 30 hours for its active metabolite roflumilast N-oxide [see Clinical Pharmacology (12.3)].
An additional placebo-controlled 1-year trial (Trial 9) evaluated the effect of DALIRESP 500 mcg on COPD exacerbations when added to a fixed-dose combination (FDC) product containing an inhaled corticosteroid and long-acting beta agonist (ICS/LABA). At screening, patients were required to have two or more exacerbations in the previous year. This trial randomized a total of 2354 patients (1178 randomized to DALIRESP, 1176 to placebo). Approximately 60% of the patients enrolled had severe COPD (postbronchodilator FEV1 30%-50% of predicted) associated with chronic bronchitis and 39% had very severe COPD (postbronchodilator FEV1 ≤ 30% of predicted) associated with chronic bronchitis; mean age of 64 years, 69% male, and 80% Caucasian. The use of long-acting muscarinic antagonists was allowed.
Effect on Exacerbations
The effect of DALIRESP 500 mcg once daily on COPD exacerbations was evaluated in five 1-year trials (Trials 3, 4, 5, 6 and 9).
Two of the trials (Trials 3 and 4) conducted initially enrolled a population of patients with severe COPD (FEV1 ≤50% of predicted) inclusive of those with chronic bronchitis and/or emphysema who had a history of smoking of at least 10 pack years. Inhaled corticosteroids were allowed as concomitant medications and used in 61% of both DALIRESP and placebo-treated patients and short-acting beta agonists were allowed as rescue therapy. The use of long-acting beta agonists, long-acting anti-muscarinics, and theophylline were prohibited. The rate of moderate or severe COPD exacerbations was a co-primary endpoint in both trials. There was not a symptomatic definition of exacerbation in these 2 trials. Exacerbations were defined in terms of severity requiring treatment with a moderate exacerbation defined as treatment with systemic glucocorticosteroids in Trial 3 or systemic glucocorticosteroids and/or antibiotics in Trial 4 and a severe exacerbation defined as requiring hospitalizations and/or leading to death in Trial 3 or requiring hospitalization in Trial 4. The trials randomized 1176 patients (567 on DALIRESP) in Trial 3 and 1514 patients (760 on DALIRESP) in Trial 4. Both trials failed to demonstrate a significant reduction in the rate of COPD exacerbations.
Exploratory analyses of the results of Trials 3 and 4 identified a subpopulation of patients with severe COPD associated with chronic bronchitis and COPD exacerbations within the previous year that appeared to demonstrate a better response in the reduction of the rate of COPD exacerbations compared to the overall population. As a result, two subsequent trials (Trial 5 and Trial 6) were conducted that enrolled patients with severe COPD but associated with chronic bronchitis, at least one COPD exacerbation in the previous year, and at least a 20 pack-year smoking history. In these trials, long-acting beta agonists and short-acting anti-muscarinics were allowed and were used by 44% and 35% of patients treated with DALIRESP and 45% and 37% of patients treated with placebo, respectively. The use of inhaled corticosteroids was prohibited. As in trials 3 and 4, the rate of moderate exacerbations (defined as requiring intervention with systemic glucocorticosteroids) or severe exacerbations (defined as leading to hospitalization and/or to death) was a co-primary endpoint.
Trial 5 randomized a total of 1525 patients (765 on DALIRESP) and Trial 6 randomized a total of 1571 patients (772 on DALIRESP). In both trials, DALIRESP 500 mcg once daily demonstrated a significant reduction in the rate of moderate or severe exacerbations compared to placebo (Table 2). These two trials provide the evidence to support the use of DALIRESP for the reduction of COPD exacerbations.
|
Study |
Exacerbations Per Patient-Year | |||||
|
DALIRESP |
Placebo |
Absolute Reduction*
|
RR† |
95% CI |
Percent Reduction‡ |
|
|
Trial 5 |
1.1 |
1.3 |
0.2 |
0.85 |
0.74, 0.98 |
15 |
|
Trial 6 |
1.2 |
1.5 |
0.3 |
0.82 |
0.71, 0.94 |
18 |
For patients in Trials 5 and 6 who received concomitant long-acting beta agonists or short-acting anti-muscarinics, reduction of moderate or severe exacerbations with DALIRESP was similar to that observed for the overall populations of the two trials.
In Trial 9, when added to background therapy of FDC ICS/LABA, the rate ratio for COPD exacerbations among patients administered DALIRESP vs. placebo was 0.92 (95% CI 0.81, 1.04).
Effect on Lung Function
While DALIRESP is not a bronchodilator, all 1-year trials (Trials 3, 4, 5, and 6) evaluated the effect of DALIRESP on lung function as determined by the difference in FEV1 between DALIRESP and placebo-treated patients (pre-bronchodilator FEV1 measured prior to study drug administration in three of the trials and post-bronchodilator FEV1 measured 30 minutes after administration of 4 puffs of albuterol/salbutamol in one trial) as a co-primary endpoint. In each of these trials DALIRESP 500 mcg once daily demonstrated a statistically significant improvement in FEV1 which averaged approximately 50 mL across the four trials. Table 3 shows FEV1 results from Trials 5 and 6 which had demonstrated a significant reduction in COPD exacerbations.
|
||||
|
Study |
Change in FEV1 from Baseline, mL |
|||
|
Trial 5 |
DALIRESP |
Placebo |
Effect* |
95% CI |
|
46 |
8 |
39 |
18, 60 |
|
|
Trial 6 |
33 |
-25 |
58 |
41, 75 |
Lung function was also evaluated in two 6-month trials (Trials 7 and 8) to assess the effect of DALIRESP when administered as add-on therapy to treatment with a long-acting beta agonist or a long-acting anti-muscarinic. These trials were conducted in a different population of COPD patients [moderate to severe COPD (FEV1 40 to 70% of predicted) without a requirement for chronic bronchitis or frequent history of exacerbations] from that for which efficacy in reduction of exacerbations has been demonstrated and provide safety support to the DALIRESP COPD program.
Starting dose titration trial
The tolerability of DALIRESP was evaluated in a 12-week randomized, double-blind, parallel group trial in patients with severe COPD associated with chronic bronchitis (Trial 10). At screening, patients were required to have had at least one exacerbation in the previous year. A total of 1323 patients were randomized to receive DALIRESP 500 mcg once a day for 12 weeks (n=443), DALIRESP 500 mcg every other day for 4 weeks followed by DALIRESP 500 mcg once a day for 8 weeks (n=439), or DALIRESP 250 mcg once a day for 4 weeks followed by DALIRESP 500 mcg once a day for 8 weeks (n=441).
Over the 12-week study period, the percentage of patients discontinuing treatment was 6.2% lower in patients initially receiving DALIRESP 250 mcg daily for 4 weeks followed by DALIRESP 500 mcg daily for 8 weeks (18.4%) compared to those receiving DALIRESP 500 mcg daily for 12 weeks (24.6%) (Odds Ratio = 0.66; 95% CI: 0.47 to 0.93; p=0.017). Because this trial was limited to 12 weeks in duration, whether initiation of dosing with DALIRESP 250 mcg improves the long term tolerability of DALIRESP 500 mcg has not been determined.
16. How is Daliresp supplied
16.1 How Supplied
DALIRESP 250 mcg is supplied as white to off-white, round tablets, embossed with “D” on one side and “250” on the other side.
DALIRESP 250 mcg tablets are available:
Blister pack 28: NDC 0310-0088-28
2X10 Unit Dose: NDC 0310-0088-39
DALIRESP 500 mcg is supplied as white to off-white, round tablets, embossed with “D” on one side and “500” on the other side.
DALIRESP 500 mcg tablets are available:
Bottles of 30: NDC 0310-0095-30
Bottles of 90: NDC 0310-0095-90
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
- Bronchospasm
DALIRESP is not a bronchodilator and should not be used for immediate relief of breathing problems (i.e., as a rescue medication).
- Psychiatric Events including Suicidality
Treatment with DALIRESP is associated with an increase in psychiatric adverse reactions. Cases of suicidal ideation and behavior, including completed suicide, have been observed in the post-marketing setting in patients with or without a history of depression.
The risks and benefits of treatment with DALIRESP in patients with a history of depression and/or suicidal thoughts or behavior should be carefully considered. Advise patients, caregivers, and families to be alert for the emergence or worsening of insomnia, anxiety, depression, suicidal thoughts or other mood changes, and if such changes occur to contact their healthcare provider so that the risks and benefits of continuing treatment with DALIRESP may be considered [see Warnings and Precautions (5.2)].
- Weight Decrease
Weight loss was a common adverse reaction in DALIRESP clinical trials. During follow-up after treatment discontinuation, the majority of patients with weight loss regained some of the weight they had lost while receiving DALIRESP. Advise patients treated with DALIRESP to have their weight monitored regularly. If unexplained weight loss occurs, patients should inform their healthcare provider so that the weight loss can be evaluated, as discontinuation of DALIRESP may need to be considered [see Warnings and Precautions (5.3)].
- Drug Interactions
The use of cytochrome P450 enzyme inducers resulted in a reduction in exposure which may result in decreased therapeutic effectiveness of DALIRESP. The use of strong cytochrome P450 enzyme inducers (e.g., rifampicin, phenobarbital, carbamazepine, phenytoin) with DALIRESP is not recommended [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
Distributed by:
AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
Daliresp® is a registered trademark of the AstraZeneca group of companies.
© AstraZeneca 2020
MEDICATION GUIDE
DALIRESP® (da'-li-resp)
(roflumilast)
Tablets
Read this Medication Guide before you start taking DALIRESP® and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment.
What is the most important information I should know about DALIRESP?
DALIRESP can cause serious side effects. Tell your healthcare provider right away if you have any of the symptoms listed below while taking DALIRESP.
- 1.
-
DALIRESP may cause mental health problems including suicidal thoughts and behavior. Some people taking DALIRESP may develop mood or behavior problems including:
- •
- thoughts of suicide or dying
- •
- attempt to commit suicide
- •
- trouble sleeping (insomnia)
- •
- new or worse anxiety
- •
- new or worse depression
- •
- acting on dangerous impulses
- •
- other unusual changes in your behavior or mood
- 2.
- Weight loss. DALIRESP can cause weight loss. You should check your weight on a regular basis. You will also need to see your healthcare provider regularly to have your weight checked. If you notice that you are losing weight, call your healthcare provider. Your healthcare provider may ask you to stop taking DALIRESP if you lose too much weight.
DALIRESP may affect the way other medicines work, and other medicines may affect how DALIRESP works. Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
What is DALIRESP?
DALIRESP is a prescription medicine used in adults with severe Chronic Obstructive Pulmonary Disease (COPD) to decrease the number of flare-ups or the worsening of COPD symptoms (exacerbations).
DALIRESP is not a bronchodilator and should not be used for treating sudden breathing problems. Your healthcare provider may give you other medicine to use for sudden breathing problems.
It is not known if DALIRESP is safe and effective in children.
Who should not take DALIRESP?
Do not take DALIRESP if you:
- •
- have certain liver problems. Talk with your healthcare provider before you take DALIRESP if you have liver problems.
What should I tell my healthcare provider before taking DALIRESP?
Before you take DALIRESP, tell your healthcare provider if you:
- •
- have or have had a history of mental health problems including depression and suicidal behavior.
- •
- have liver problems
- •
- have any other medical conditions
- •
- are pregnant or plan to become pregnant. It is not known if DALIRESP will harm your unborn baby. Talk to your healthcare provider if you are pregnant or plan to become pregnant.
- •
- are breastfeeding or plan to breastfeed. It is not known if DALIRESP passes into your breast milk. You and your healthcare provider should decide if you will take DALIRESP or breastfeed. You should not do both.
How should I take DALIRESP?
- •
- Take DALIRESP exactly as your healthcare provider tells you to take it.
- •
- DALIRESP can be taken with or without food.
- •
- If you take more than your prescribed dose of DALIRESP, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of DALIRESP?
DALIRESP can cause serious side effects, including:
See “What is the most important information I should know about DALIRESP?”
The most common side effects of DALIRESP include:
- •
- diarrhea
- •
- weight loss
- •
- nausea
- •
- headache
- •
- back pain
- •
- flu like symptoms
- •
- problems sleeping (insomnia)
- •
- dizziness
- •
- decreased appetite
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of DALIRESP.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How do I store DALIRESP Tablets?
- •
- Store DALIRESP at 68°F to 77°F (20°C to 25°C); excursions permitted to 15° - 30°C (59° - 86°F). [See USP Controlled Room Temperature].
Keep DALIRESP Tablets and all medicines out of the reach of children.
General information about DALIRESP
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use DALIRESP for a condition for which it was not prescribed. Do not give DALIRESP to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about DALIRESP. For more information about DALIRESP, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about DALIRESP that is written for health professionals.
For more information about DALIRESP call 1-800-236-9933.
What are the ingredients in DALIRESP?
Active ingredient: roflumilast
Inactive ingredients: lactose monohydrate, corn starch, povidone and magnesium stearate.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Rev. 04/2017
Distributed by:
AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850


| DALIRESP
roflumilast tablet |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| DALIRESP
roflumilast tablet |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - AstraZeneca Pharmaceuticals LP (054743190) |
| Registrant - AstraZeneca (230790719) |
More about Daliresp (roflumilast)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (63)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- FDA approval history
- Drug class: selective phosphodiesterase-4 inhibitors
- Breastfeeding
- En español
